
http://www.ted.com/talks/lang/spa/seth_berkley_hiv_and_flu_the_vaccine_strategy.html
De: Jorge Chirinos <jchirinosmd@yahoo.es>
Para: SALUD_LORETO@yahoogroups.com
Enviado: dom,20 junio, 2010 21:40
Asunto: Re: [SALUD_LORETO] Quien es el Dr Juan Gérvas?????????
http://www.cgcom. org/search/ node/Juan Gervas
http://www.cgcom. org/search/ node/8 razones para no vacunarse
Lo he encontrado en youtube:
http://www.youtube. com/watch? v=WisgpB_ le1w
http://www.youtube. com/watch? v=lB8K6RQjQ3A&NR=1
http://www.youtube. com/watch? v=WsQUrCVMPuw&NR=1
http://www.youtube. com/watch? v=tQzj4V0ledk&feature=related
http://www.youtube. com/watch? v=QVdPG8jYD- c
Por ejemplo, dice que la efectividad de las vacunas estacionales en ninhos es del 33%, y calla que la eficacia es del 82%. (http://www2. cochrane. org/reviews/ en/ab004879. html). Efectividad en este caso es disminucion del numero de sintomas gripales y eficacia es disminucion de los casos confirmados de influenza. (http://www.uc. cl/medicina/ medicinafamiliar /html/articulos/ 215.html)
Donde si he encontrado referencias sobre la inmunidad y Influenza A es en Medscape (ver mas abajo).
Ni Wikipedia ni los blogs que no contienen referencias pueden ser fundamento para tomar decisiones clínicas. Desgraciadamente el Dr. Gervas no especifica CON EVIDENCIAS y cientificamente si debe vacunar o no. Filosofa en base a principios y generalidades, hace juegos de logica y de alli llega a conclusiones sin probar de manera cientifica ninguna. Asi llega a la conclusion que no hay que prevenir, estando en contra de vacunas, de mamografias etc.
Saludos,
Jorge Chirinos
http://www.medscape .com/viewarticle /580490
Innate Immunity to Influenza Virus: Implications for future therapy.
Summary and Introduction
Summary
Innate immunity is critical in the early containment of influenza virus infection. The innate response is surprisingly complex. A variety of soluble innate inhibitors in respiratory secretions provide an initial barrier to infection. Dendritic cells, phagocytes and natural killer cells mediate viral clearance and promote further innate and adaptive responses. Toll-like receptors 3 and 7 and cytoplasmic RNA sensors are critical for activating these responses. In general, the innate response restricts viral replication without injuring the lung; however, the 1918 pandemic and H5N1 strains cause more profound, possibly harmful, innate responses. In this review, we discuss the implications of burgeoning knowledge of innate immunity for therapy of influenza.
Introduction
Influenza virus remains a very important threat to global health, causing approximately 36,000 deaths annually in the USA alone.[1] Although in nonpandemic years the virus may be considered seasonal and primarily affecting certain age groups (the elderly and children), the pandemic of 1918 caused by the H1N1 subtype resulted in millions of fatalities including those of healthy, young adults.[2] There is currently major concern about the potential for a new pandemic arising from avian strains such as those of the H5N1 subtype.
Innate immunity refers to host defense functions that are directly encoded in the genome and are capable of recognizing and inhibiting infectious agents or transformed host cells. Recent studies have revealed surprising complexity and specificity for innate immune mediators, a prominent example being the Toll-like receptor (TLR) family.[3] In addition to providing a first line of host defense against infection, innate immunity strongly contributes to promotion and direction of the adaptive immune response. For many infections, innate immunity provides a vital bridge between first exposure of the naive host and subsequent elaboration of specific antibody and T-cell responses. Influenza viral infections provide a prime example of the importance of innate immunity because this virus is able to undergo continuous genetic variation, allowing it to evade prior adaptive immune responses. This review will focus on the mechanisms through which the host protects itself from death or severe morbidity during the first days after infection with a novel influenza viral strain.
After initial exposure to a novel influenza viral strain, it takes between 5 and 7 days before specific antibodies and T cells arrive in the lung to definitively clear the virus; hence, this defines the time window in which innate immunity is critical. In most cases, influenza viruses remain confined to the upper respiratory tract in humans, despite the ability of the virus to bind to most cells. Innate immune mechanisms are critical in restricting the anatomic spread of influenza virus and facilitating the rapid development of adaptive responses. Clearly, the innate response is not yet fully understood. One important controversy is the extent to which some aspects of the innate response may contribute to morbidity, or even mortality, through inflammation. Overall, the optimal outcome would be that the innate response prevents dissemination of the virus without harmful inflammation and promotes effective adaptive responses that can clear the infection definitively and prevent future infections with the same viral strain (or even better, multiple viral strains). Since we cannot do full justice to the increasingly complex topic of innate immunity to influenza virus in one review, we emphasize certain areas (e.g., soluble innate inhibitors, which are our main research interest) and reference key reviews for other important areas (e.g., type 1 interferons or dendritic cells [DCs]) that we describe more briefly.
Innate Barriers to Infection in the Respiratory Tract
This review will mainly focus on innate defense in the upper and lower respiratory tract since these are the major target sites for influenza viral infection. It should be noted briefly, however, that initial barriers to infection also occur in the oral cavity and possibly even in tears. For instance, saliva has strong influenza viral-neutralizing activity that is partly provided by salivary agglutinin (also called scavenger receptor cysteine-rich glycoprotein [gp]-340), as well as by mucins and other components.[4,5] The eye is a possible portal of entry for influenza viruses and tears contain surfactant protein (SP)-D, which can inhibit influenza.[6,7]
One can view the innate immune response to viral infection as proceeding in different temporal phases, including: initial inhibition by soluble proteins present in mucosal secretions prior to infection, detection of infection mediated by soluble or cellular sensors of infection, deployment of effectors to limit infection and prevent dissemination, and recruitment of innate and adaptive effectors to lead to resolution of infection and reduction of inflammation. As with other methods of analyzing a complex biological response, this approach is oversimplified since there is overlap between these phases.
Soluble Effectors of Innate Immunity
Inhibitors of Influenza Present in Respiratory Lining Fluid Prior to Infection
Mucins, gp-340 & Pentraxins. Mucins, gp-340 and pentraxins are present in bronchoalveolar lavage fluid (BALF) and inhibit influenza virus by providing decoy sialic acid ligands to which the virus binds.[5,8] The effectiveness of these inhibitors against human- or avian-like strains of influenza depends on the amount of α(2,3)- versus α(2,6)-linked sialic acids they display. Oseltamivir potentiates the antiviral activity of mucins, presumably by preventing the neuraminidase (NA) from freeing the viral hemagglutinin (HA) bound to mucin.[9] Mucins and gp-340 do not increase neutrophil uptake or respiratory burst responses to influenza virus.[10] Mucin and gp-340 induce viral aggregation and may promote viral clearance through mucociliary mechanisms, although this has not yet been demonstrated.
Collectins. Collectins are a family of collagenous lectin molecules present in blood and mucosal (including respiratory) secretions that are able to recognize distinctive patterns of carbohydrates decorating the surface of viruses, bacteria, fungi and protozoa.[11,12] The basic structural unit of collectins is a trimer composed of a disulfide containing a N-terminus, a structurally important collagen domain, a trimerizing neck domain and a carbohydrate- recognition domain (CRD) that mediates attachment to carbohydrates. In general, collectins form higher-order multimers through disulfide bonding between trimers at the N-terminus. Collectins also bind to inflammatory cells and can modulate activation of these cells, resulting in either an increase or decrease in activation.[10] Our laboratory has studied the interactions of collectins with influenza virus extensively so we will provide a fairly comprehensive discussion of their antiviral activities. Collectins bind to and directly neutralize influenza virus.[13] The collectins appear to play important roles in restricting influenza replication in the early phase of infection and in preventing excessive inflammation. There are at least three collectins in humans; two were originally isolated from pulmonary fluids, SP-D and SP-A, and another from serum, mannose-binding lectin (MBL). Recent findings indicate that SP-D is not only present in the lung but also in a variety of mucosal or other epithelial surfaces in the body.[14]
SP-D & MBL. The first evidence of the role of collectins in immunity against influenza virus was the observation that mammalian serum β-inhibitors of the virus are collectins. SP-D (and to a lesser extent SP-A) was then shown to inhibit infectivity of influenza virus and to contribute strongly to the antiviral activity of BALF.[13] SP-D-knockout (SP-D -/-) mice show increased viral titers, weight loss and inflammatory responses after influenza infection[15,16] These effects can be corrected by instillation or overexpression of wild type or mutant forms of SP-D in the lung.[15,17,18] Sensitivity of various influenza viral strains depends on the level of glycosylation of the envelope proteins, especially the HA; strains lacking high mannose oligosaccharides on the globular head of the HA are resistant to SP-D or MBL. Such strains generally have an increased ability to replicate in mouse lungs and cause increased illness in mice (e.g., increased weight loss or even mortality)[19,20] and their replication is not altered in SP-D -/- mice compared with controls.[16] Of note, the mouse-adapted A/PR/8 strain that is used in many murine studies of influenza infection has no high mannose oligosaccharides on its envelope proteins and is resistant to SP-D or MBL.[21] In effect, such studies characterize the immune response occurring in the absence of a direct contribution of SP-D.
It is of interest that pandemic strains of influenza A virus, including those isolated in 1918 (H1N1), 1968 (H3N2) and 1977 (reintroduction of H1N1), have fewer glycan attachments on the HA globular head and acquire increased glycans as they evolved in the human population.[22] In the case of the reintroduced H1N1 strain and H3N2 strains, the initial pandemic strains were resistant to SP-D but later strains that had more glycosylation were highly sensitive.[19] Based on knowledge of glycosylation of the 1918 strain, it is likely that it too is resistant to SP-D.[23] Human isolates of H5N1 also have reduced glycosylation on the HA head compared with recent human H3N2 or H1N1 strains and are resistant to SP-D in vitro (Hartshorn et al., Unpublished Data). Hence, glycosylation of the HA head appears to confer adaptive advantages to influenza viral strains after introduction into the human population, while also rendering the viruses more sensitive to collectin-mediated inhibition. This suggests that there is a trade off in which added glycosylation protects the HA from antibody-mediated neutralization[24] and possibly has other beneficial effects (e.g., reducing binding affinity of the HA, which makes the virus less dependent on NA activity),[25] while possibly attenuating disease severity through increased sensitivity to collectins. This sort of adaptive evolution of human influenza viral strains may be subtype dependent because the H2N2 subtype was limited in its ability to add glycans on the HA head without compromising HA-binding affinity and fusion activity to such an extent that infectivity was diminished.[26] This limitation is postulated to contribute to the relatively brief tenure of H2N2 in the human population (i.e., because it lacked flexibility in masking its HA from antibody recognition) . Overall, these findings also suggest that lack of HA glycosylation of pandemic strains may be one factor allowing more rapid spread or greater virulence, since they evade inhibition by SP-D or MBL, but that subsequent adaptive glycosylation may be an important mechanism of survival in the human population.
Various clinical findings also indicate a protective role for SP-D against influenza virus. Some conditions are associated with acquired deficiency of SP-D, including cystic fibrosis (CF), chronic smoking and chronic obstructive pulmonary disease (COPD).[27-29] These conditions also predispose to influenza viral infection. Neutrophilic inflammation in the lung can result in degradation of SP-D through action of neutrophil proteases, leading to increased susceptibility to infection.[30] Diabetes mellitus also predisposes to severe influenza virus infection. In a mouse model of diabetes, increased severity of influenza was linked to inhibition of host defense activity of SP-D by elevated glucose.[31] There are polymorphic forms of SP-D that differ in their level of multimerization in vivo and also differ in anti-influenza activity.[32] SP-D polymorphisms have been associated with susceptibility to respiratory syncytial virus (RSV) or Mycobacteria tuberculosis infection,[33,34] but an association with influenza incidence or severity has not yet been studied. There are polymorphisms of MBL associated with markedly reduced levels of the protein in the blood and increased risk for infections of various kinds, including respiratory viral infections;[35] hence, MBL may play a role in host defense against influenza virus.
SP-A. The mechanism of anti-influenza activity of SP-A differs from that of the other collectins; instead of binding to glycans on the viral HA and NA, the viral HA binds to a highly sialylated glycan present on the CRD of SP-A.[13] Hence, SP-A inhibits influenza virus through a mechanism similar to that of mucins, gp-340 or pentraxins. SP-A is present in higher concentrations in respiratory lining fluid than SP-D and contributes to host defense against influenza, RSV and various bacteria.[36] Studies involving human BALF and SP-A -/- and/or SP-D -/- mice indicate that SP-A has a less important role than SP-D in the innate response to influenza viral infection. However, consistent with its different mechanism of action, SP-A has activity against influenza viral strains resistant to SP-D so it may provide a level of protection against pandemic strains or other strains lacking glycans on the HA head.[37] Of interest, gp-340 and pentraxins also inhibit strains that are not inhibited by SP-D.
Porcine SP-D. Although SP-D of most species (and all of the serum collectins) lack glycan attachments on the CRD, this is not true of porcine SP-D, which has a glycan attachment on the lateral surface of the CRD (see Figure 1 for a picture of the neck and CRD or neck and carbohydrate recognition domain [NCRD] of SP-D showing the location of the glycan on porcine SP-D).[38] This enables porcine SP-D to inhibit influenza virus via a dual mechanism that involves both CRD binding to high mannose glycans on the virus and binding of the viral HA to sialic acids on porcine SP-D. As in the case of mucins and gp-340, the ability of SP-A or porcine SP-D to inhibit avian-like strains of influenza A virus is determined by the density of α(2,3)-linked sialic acids on SP-A or porcine SP-D, and the deliberate addition of α(2,3)-linked sialic acids in vitro on porcine SP-D caused greatly increased inhibition of the A/PR/8 strain that binds preferentially to this linkage.[38]
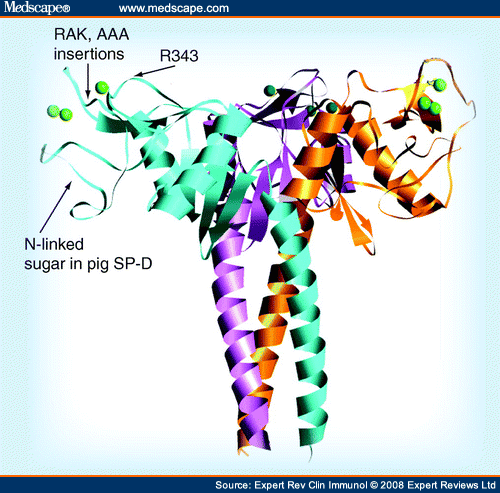
Figure 1. Diagram of the neck and carbohydrate recognition domains of SP-D. This ribbon diagram was derived from x-ray crystallographic studies of the trimeric binding domain of SP-D in association with the coiled coil neck domain, which is responsible for trimerization. Three saccharide-binding pockets are located on the flat upper surface of the trimer (see spherical single green calcium ion in the pocket). The binding pocket is surrounded on either side by ridges, and substitution of the R343 residue that forms one of the ridges can greatly increase influenza viral-neutralizing activity. Insertions of amino acids (e.g., RAK or AAA, as shown) adjacent to the other ridge also increases antiviral activity to a lesser extent. The location of the N-linked sialylated glycan on pig SP-D is also shown. SP = Surfactant protein.
Natural IgM. Natural IgM (nIgM) is a form of IgM that is largely produced by B1 cells in the absence of class-switch recombination and somatic hypermutation and contributes to innate defense against various infections, including influenza. B1 cells are a distinct population of B cells that differ from B2 cells in their location and means of activation. B2 cells are classic B cells activated by antigen exposure to proliferate and undergo clonal expansion, leading to the generation of high-affinity antigen-specific antibodies. B1 cells produce nIgM reactive with non-T-cell-dependen t antigens present on various pathogens and some self antigens. B1 and B2 cells have been shown to be separately regulated in response to influenza in mice in elegant studies by Baumgarth et al..[39] B1 cells are the predominant source of nIgM, which has influenza virus-inhibiting activity, is present in mice in the absence of prior influenza exposure and does not rise after viral exposure. Virus-specific IgM produced by B2 cells begins to rise at approximately day 7 and remains elevated through to day 21. Murine nIgM has been shown to mediate influenza viral neutralization in vitro through fixation of complement on viral particles through the classical pathway, leading to viral aggregation and inhibition of HA binding.[40] Hence, although IgA produced as part of the adaptive immune response is critical for protection against recurrent infection based on its expression in mucosal surfaces, nIgM may play a role in the early phase of defense against infection with novel influenza viral strains.
Complement. Complement components are present in alveolar lining fluid.[41] Complement may participate in the innate response to influenza virus by nIgM through the classical pathway of complement activation. Complement can also be deposited on influenza viral particles in the presence of MBL through the 'lectin pathway' of complement activation. MBL structurally resembles C1q and binds to specific MBL-associated serum proteases that lead to complement activation and deposition on targets.[42] Although MBL is principally a serum protein, measurable levels are found in the lung during influenza viral infection and it can trigger complement-dependen t viral neutralization in vitro .[19,43] The role of MBL in vivo in influenza viral infection is under investigation. The ability of MBL to promote complement activation is not shared by other collectins and, in fact, SP-A inhibits complement activation in the lung;[41] hence, it is possible that MBL has distinctive roles in influenza infection compared with SP-D and SP-A. The role of complement in innate defense against influenza virus in vivo has not yet been studied extensively; however, C3 and C5 have been shown to play important roles in T-cell priming and recruitment to the lung and generation of specific CD8+ T-cell responses.[44,45]
Defensins. Defensins are a family of cationic antimicrobial peptides that have activity against a wide range of bacterial, yeast and enveloped viruses.[46-49] α-defensins are produced by human neutrophils (called human neutrophil peptides [HNPs]), are stored in primary granules of these cells and are released in large quantities in response to activating stimuli. Additional α-defensins are released by intestinal epithelial Paneth cells and other immune cells also produce α-defensins. β-defensins are produced by various epithelial cells (including those in the respiratory epithelium). A variety of other defensins and related antimicrobial peptides are produced in humans and many other animal species. It is not yet clear the extent to which defensins contribute to host defense against influenza in vivo, although the concentrations of HNPs present in saliva or BALF treated with activated neutrophils are sufficient to mediate inhibition.[30] As with collectins, defensins have broad-spectrum antimicrobial activity and lectin activity; however, HNPs have activity against the A/PR/8 strain that is resistant to SP-D or MBL.[49] HNPs can also cause viral aggregation and promote uptake of influenza by neutrophils.[50] Defensins have a variety of other effects, including chemotaxis for T cells and DCs and stimulation of cytokine generation.[46] We have found that HNPs bind to SP-D and can either interfere with or, more rarely, increase antiviral activity of purified recombinant SP-D or natural SP-D in BALF.[30,32,49] The ability of HNPs to bind to and precipitate SP-D from BALF may account, in part, for reduced levels of SP-D in some chronic inflammatory states, such as CF or COPD.
Sensors of Infection in the Respiratory Tract
TLRs and the Retinoic Acid-inducible Gene 1
Our understanding of the mechanisms through which mammalian cells sense the presence of viral infection and initiate innate responses has expanded greatly in recent years.[3,51] Innate recognition of viral RNA is critical in triggering type 1 interferon (i.e., IFN-α/β) responses that are in turn critical for initial containment of influenza virus infection (see later). There are two major viral RNA-recognition systems involving TLRs as well as a cytoplasmic- recognition system (mediated by RIG-I or melanoma-differenti ation antigen [MDA]-5).
The TLR system involves recognition of viral RNA in endosomal compartments through either TLR3 or TLR7. TLR3 principally recognizes dsRNA, while TLR7 recognizes ssRNA. These pathways are particularly important for recognition of RNA viruses that enter the cell through endocytic pathways (e.g., influenza virus). TLR7 has been shown to be important in triggering recognition of viral ssRNA, leading to robust induction of type 1 interferons by plasmacytoid DCs (pDCs).[52] TLR7 activation is linked to the process of influenza viral fusion and uncoating in late endosomes, and interfering with these processes by inhibiting endosomal acidification inhibits TLR7 activation.[53] TLR3 participates in response to influenza virus by respiratory epithelial cells,[54,55] and expression of TLR3 in the lung is upregulated by influenza virus infection.[56]
The cytoplasmic RNA-recognition system, specifically mediated by RIG-I, is also importantly involved in triggering innate responses to influenza virus. Although RIG-I was initially identified as a helicase that specifically recognizes dsRNA, recent papers demonstrate that it recognizes 5´-capped ssRNA of the influenza virus.[57] Of interest, this recognition can be blocked by the influenza viral NS1 protein. The RIG-I pathway leads to activation type 1 interferon release via the mitochondrial antiviral signaling protein (MAVS) that is coupled to mitochondria and then via nuclear factor (NF)-κB and interferon-regulato ry factor (IRF)3. There is some specificity to the cytoplasmic RNA-sensing pathways, in that the other major sensor (MDA5) is involved in recognition of other RNA viruses, whereas RIG-I is essential for response to influenza virus.[58,59] MAVS -/- mice develop normally but exhibit significantly impaired host defense against some RNA viruses (e.g., vesicular stomatitis virus)[60] Recent studies confirm that influenza virus also generates type I interferon production through both RIG-I and TLR7 pathways and that use of these pathways are cell-type specific (RIG-I in epithelial cells and conventional DCs; TLR7 in pDCs and B cells).[52,61,62]
Complex Role of NF-κB in the Influenza Virus Life Cycle and Innate Immune Response to the Virus
The NF-κB signaling pathway is activated in many cell types by influenza viral infection and it, in turn, regulates the expression of key proinflammatory cytokines, including IL-1, IL-6, IL-8 and TNF-α. Activation of NF-κB can be mediated by activation of the IκB kinase that phosphorylates IκB, targeting it for degradation. Since IκB constitutively suppresses NF-κB activation, loss of IκB promotes NF-κB activation. Initially, it was believed that this signaling pathway was part of a protective immune response to influenza; however, a variety of recent studies indicate that activation of NF-κB can promote influenza viral replication in respiratory epithelial cells or Epstein-Barr virus-transformed B cells.[63] The mechanism for enhanced viral growth was found to involve promotion of viral ribonucleoprotein (RNP) export from the nucleus through activation of caspases in the early phase of apoptosis.[64] NF-κB has complex effects that appear to vary depending on the cell type or species of virus (or even on the influenza viral subtype) under consideration. From these findings, it would appear that influenza viruses can take advantage of an innate immune signaling pathway to favor their own growth. Viruses such as HIV or HSV do a similar thing by containing NF-κB-regulated motifs in their promoters.
Other studies, however, indicate that the relationship of influenza with NF-κB signaling may be more complicated. A study by Bernasconi et al. using A549 cells and the mouse-adapted WSN H1N1 influenza viral strain found that NF-κB activation was not essential for viral replication but that it did mediate IL-8 generation.[65] Thus, in different experimental models, viral activation of NF-κB has been found to be deleterious through direct promotion of viral replication or through promotion of deleterious inflammation. A recent study may, in part, reconcile some of these findings. Wei et al. found that fibroblasts lacking NF-κB showed increased upregulation of interferon-stimulat ed genes (ISGs) and increased sensitivity to the antiviral effects of type 1 interferon against influenza virus.[66] NF-κB was shown to negatively regulate expression of ISGs by binding to their promoters. Hence, NF-κB may promote viral replication through antagonizing the response to interferon. Of interest, NF-κB and interferon signaling pathways are both activated by TLR7/MyD88 through shared and unique intermediates, and the relative extent to which interferon or NF-κB is activated depends on the cell type.[53] NF-κB and interferon pathways are subject to negative regulation by suppressor of cytokine signaling (SOCS)3 during influenza viral infection of respiratory epithelial cells.[67] Of interest, SOCS1 is also upregulated by influenza viral infection, but this protein suppresses the interferon promoter without suppressing NF-κB, implying differential regulation of these pathways.
MAPK and PKC Signaling and Influenza
Influenza virus has a complex life cycle that involves uptake into endosomes and trafficking to late endosomes in the endosomal-lysosomal pathway, with release of the viral core into the cytoplasm of the cell after activation of the fusion domain of the HA at low pH. Viral RNPs are released from attachment to the M1 protein through the action of the M2 channel protein that mediates acidification of the viral interior. The RNPs are then transported to the nucleus of the cell where RNA transcription occurs. Newly formed RNPs are then transported out of the nucleus for assembly into new particles. Viral MAPK and PKC activition in infected cells also has an important role in the viral life cycle. PKC has at least 11 isoforms that may serve different functions. In the case of influenza infection, a PKCβ isoform has been found to mediate viral RNP transfer into the nucleus. By contrast, PKCα has been found to contribute to export of the newly formed RNPs from the nucleus. Recently, this PKCα activation was shown to be mediated by MAPK activation resulting from assembly of newly formed HA proteins in raft domains on the cell surface.[68] This appears to coordinate RNP export at a time when envelope proteins are assembled and ready for packaging of new viral particles. Inhibition of PKC has been shown to inhibit influenza viral replication and some proinflammatory actions of the virus.[69-71]
Type 1 Interferons
It is clear from this discussion that one key outcome of initial infection is the generation of type 1 interferons. Type I interferons (IFN-α and IFN-β) are an important part of host defense against influenza and many other viruses. They induce an antiviral state in cells and also potentiate responses of innate and adaptive immune effector cells. We will not be able to fully describe the multiple antiviral activities of type 1 interferons but many useful reviews are available.[72] Deletion of key interferon signaling proteins or receptors for IFN-α/β in mice results in greater mortality after infection with a highly virulent influenza virus strain, accompanied with systemic (as opposed to respiratory- restricted) viral infection.[73] Of note, however, such mice do not suffer worse infection with less-virulent influenza viral strains.[74] One key feature of influenza viruses is that they induce a comparatively blunted type 1 interferon response; influenza viruses have evolved means of counteracting type 1 interferons. The NS1 protein of influenza viruses inhibits IFN-β production in infected epithelial cells.[75] This inhibition involves binding of dsRNA by the NS1 protein and interactions of NS1 with RIG-I. Deletion of the NS1 protein prevents growth of the virus except in IFN-α/β-deficient systems. Gene array analysis on influenza virus-infected respiratory epithelial cells demonstrate that deletion of the NS1 protein results in greater induction of genes in interferon, NF-κB and other antiviral pathways.[76] Of note, the NS1 gene of the 1918 pandemic virus is particularly effective at inhibiting antiviral response genes, implying that the increased pathogenicity of the 1918 strain might have resulted from more effective inhibition of innate antiviral responses. Influenza viruses have other less-well-character ized means of blunting interferon responses apart from NS1.[72] Respiratory epithelial cells are an important source of type 1 interferon production early in infection and pDCs produce large amounts of type 1 interferon subsequently.[77]
Chemokines and Cytokines
Although space does not allow for a full discussion of this important topic, it must be noted that respiratory tract epithelial cells and immune responder cells also produce abundant amounts of various cytokines and chemokines that result in some of the symptoms of influenza but also play a role in mounting an inflammatory response that contributes to clearance of infection.[78-83] More references related to this topic are noted in the next section.
Cellular Effectors in the Innate Immune Response
Respiratory Epithelial Cells
Upper respiratory tract epithelial cells are the first target cells for influenza virus infection and the first cells to mount an innate response. Binding of influenza viruses to respiratory epithelial cells, as to other cells, is mediated by attachment of the HA to sialic acids expressed on proteins or lipids on the surface of the cells.[54,55,84,85] Human and avian influenza A strains differ in the tropism of their HA for either specific sialic acid linkages (i.e., avian strains selectively bind to sialic acids in an α(2,3)-linkage to galactose; whereas, human strains prefer an α(2,6) linkage)[86] This tropism causes restriction of the ability of avian strains to attach to the human upper respiratory epithelium, which largely expresses α(2,6)-linked sialic acids. Bird respiratory and gastrointestinal epithelia largely express α(2,3)-linked sialic acids, as do respiratory epithelia of mice. Of interest, the distal respiratory epithelia (e.g., that of alveoli) of humans expresses more α(2,3)-linked sialic acids, perhaps contributing to the increased propensity of H5N1 viruses to cause viral pneumonia.[87] As noted, influenza virus infection remains largely confined to the respiratory tract despite the presence of viral receptors on many cells types in the body. An important factor leading to selectivity of the virus for respiratory cells is their expression of specific proteases that are able to cleave the viral HA.[88] This cleavage is a necessary step in the infectious cycle. The HA of some avian strains is more readily cleaved by ubiquitous proteases, allowing systemic infection in animal hosts.
Infection of respiratory epithelial cells with influenza results in release of type 1 interferons and chemokines, such as IL-8, that promote recruitment of neutrophils. Influenza is a lytic infection leading to death of respiratory epithelial cells. Apoptotic and necrotic epithelial cells then release factors that promote phagocytosis[89] by macrophages or neutrophils and recruitment of neutrophils.[90] TLR3 is expressed on respiratory epithelial cells and is activated by influenza viral RNA, leading to release of inflammatory mediators.[54] The respiratory epithelium can also release defensins and surfactant proteins that can contribute to antiviral defense.[91] Hence, it is appropriate to view the respiratory epithelial cells as immune response cells.
Dendritic Cells
Dendritic cells are pivotal in mounting an effective innate and adaptive response to respiratory infections. DCs are present at baseline levels in the conducting airways, forming a network with their dendrites that can sample the airway lumen for infection. We refer readers to an excellent recent review of the role of DCs in respiratory viral infection since we cannot do full justice to this fascinating field.[92] DCs express many TLRs, enabling them to react to a broad array of pathogens or products of pathogens. DC populations are highly dynamic and their numbers in the lung are increased after viral or other infection. There are two major subtypes of DCs involved in innate response to influenza: conventional DCs (cDCs) and pDCs. cDCs are in an immature state prior to infection and undergo maturation and migration to draining lymph nodes after infection, where they participate in antigen presentation.[93] pDC numbers in the lung increase after viral infection and these cells are a major source of type 1 interferon production. The profound ability of pDCs to generate type 1 interferon appears to relate to increased constitutive expression of IRF-7.[94,95] cDCs and pDCs produce successive waves of cytokines after influenza virus infection in vitro, including early (2-4 h after infection) production of cytokines and chemokines for recruitment of natural killer (NK) cells and neutrophils and, later, production of cytokines and chemokines that attract T and B cells.[96] Infection of DCs with influenza virus also results in expression of viral HA on the DC surface, which engages NK cell receptors and this, along with DC-mediated release of type 1 interferon and IL-12, potentiates activation of NK cells.[97] Clearly, DCs contribute to the coordination of many aspects of the innate and adaptive response to influenza.
Interactions of influenza viral strains with DCs may be an important determinant of virulence. For instance, H5N1 strains can replicate in cDCs, leading to production of large amounts of virus and cell death.[98] By contrast, pDCs are protected by virtue of their strong production of type 1 interferons. The influenza virus NS1 protein counteracts DC maturation and reduces their ability to promote a Th type 1 response.[99] This effect appears to be separate from the ability of the NS1 protein to suppress type 1 interferon generation.
NK Cells
Recent studies have confirmed a very important role for NK cells in the host response to influenza virus infection and have clarified the mechanisms through which NK cells recognize and become activated in response to influenza. Gazit et al. have shown that mice lacking the NK cell receptor, NKp46, have increased mortality after influenza viral infection.[100] Recognition of influenza virus by NK cells is, in part, mediated by binding of the viral HA to NKp44 and NKp46.[97,101] This recognition provides specificity to the interaction of DCs with NK cells, leading to activation of the NK cells.[97] NKp44 binds to common human strains of influenza but also to the H5N1 subtype of avian influenza virus.[102] Recent human strains are less effective at triggering NK-mediated lysis, perhaps due to increased glycosylation of the viral HA, which can reduce binding affinity of the HA for sialylated receptors, such as NKp44 or NKp46.[103]
Macrophages
Alveolar macrophages are important both in killing infectious organisms that reach the lower airways and in release of chemokines and cytokines that have proinflammatory effects and recruit other cells to the lung. Blood monocytes or macrophages derived from blood monocytes recruited to the lung also participate in the early host response to influenza. An important beneficial activity of macrophages during influenza infection is the phagocytosis of virus-infected apoptotic cells.[89,104] Both macrophages and neutrophils participate in this mechanism of viral clearance in mouse models and the macaque model of influenza infection.[89,104] In a mouse model of infection with the WSN influenza strain, inflammation and mortality were increased by inhibition of phagocytosis. Phagocytosis of virus-infected cells also correlated with clearance of virus from the lung. Although TLR4 does not transduce signals directly in response to influenza virus, it appears to contribute to defense against influenza by promoting phagocytosis of influenza virus-infected cells.[89,105]
Influenza virus undergoes abortive infection in monocytes or neutrophils and, hence, these cells are unlikely to serve as a reservoir for influenza virus (in contrast to the case of HIV).[106,107] An important exception is that H5N1 strains can productively infect monocytes. Depletion of macrophages from mice or pigs has been shown to impair the response to influenza virus.[108,109] Infection of mice with the 1918 strain of influenza causes greatly increased neutrophil and monocyte recruitment to the lung, and depletion of these cells increases mortality.[110] Of interest, the PB1-F2 protein produced by an alternative reading frame of the PB1 polymerase gene appears to increase the pathogenicity of influenza viruses by causing apoptosis of macrophages.[111,112] Although the PB1-F2 protein is not essential for viral replication, it potentiates viral virulence in vivo. The reading frame for PB1-F2 is present in all recent pandemic strains and has been selectively preserved in the recent evolution of H5N1 strains.
In a study by Lin et al., CCR2 was shown to be critical in monocyte recruitment to the lung of influenza-infected mice (A/PR/8 strain).[113] These monocytes differentiate into monocyte-derived DCs that express NOS2 and robustly stimulate T-cell proliferation. They also give rise to exudate macrophages that express high levels of TNF-α and NOS2. These cells appear to play a largely harmful role since CCR2 -/- mice lack the monocyte influx and have increased survival without increased viral load in response to influenza infection. Overall, however, monocytes and macrophages appear to play a protective role during influenza virus infection.
Neutrophils
Neutrophils are the predominant cell recruited to the influenza virus-infected airway in the first several days after infection. This has been shown in mouse models but also in the ferret and macaque models of infection that more closely simulate human infection.[114-117] The neutrophil influx is more intense and sustained in severe influenza infection (e.g., that caused by the 1918 strain or by infection of SP-D -/- mice with recent human strains). The initial influx of neutrophils is probably triggered by specific signals released by respiratory epithelial cells and DCs. The role of the neutrophils in viral clearance or pathogenesis is not yet fully clear. As noted earlier, phagocytosis of virus-infected apoptotic cells by neutrophils and macrophages appears to play an important role in viral clearance in mouse models. Inhibition of the neutrophil influx in mice has been shown to worsen the outcome of infection in some cases but to improve the outcome in others.[118-121] Infection of CXCR2 -/- mice with influenza results in markedly reduced neutrophil recruitment, but this did not significantly alter the course of infection.[118-120]
As noted, the macaque model of influenza virus infection appears to closely parallel human infection. Infection of macaques with the Texas 91 H1N1 strain used in human experimental studies results in a clinical syndrome similar to relatively low virulence human infection with rhinorhea, with some weight loss and clearance of the virus by day 7. Infection predominates in the upper airways but focal bronchopneumonia is also present. This model has been used to develop cDNA microarray and proteomic profiles of the innate and adaptive response to influenza.[114-116] At day 4, a disproportionate number of neutrophils are found in BALF and on tissue sections of involved areas of airway and lung. These cells label with viral antigen, indicating phagocytosis of virus or virus-infected cell debris. Many genes related to neutrophil recruitment and activation are also upregulated at day 4. In the macaque model, the 1918 virus is less effectively cleared and is associated with a more intense and sustained neutrophil (and other inflammatory cell) response compared with recent human strains.[122] Similar findings have been obtained in mice infected with a recombinant 1918 strain.[123] Several genes associated with neutrophil recruitment in mice (MIP-1, MIP-2, IL-1, IL-6 and G-CSF) were also disproportionately upregulated in this model. As previously noted, blockade of neutrophil and monocyte recruitment in mice infected with the 1918 strain increases mortality, indicating that the cells play a predominantly protective role.
Influenza virus directly binds to, and is taken up by, neutrophils and these cells generate a respiratory burst in response to the virus.[70,124] Neutrophil generation of reactive oxygen species (ROS) is increased by preincubation of the virus with collectins.[13,21] Of interest, however, is that pre-incubation of neutrophils with SP-D reduces the ROS response when influenza is added later (i.e., preincubation of the virus with SP-D is needed to generate the enhanced response).[10] Although it is not currently possible to reach a firm conclusion regarding the extent to which neutrophils are protective versus harmful during influenza infection, a hypothesis consistent with the available data is that the phagocytic role of neutrophils is largely beneficial, whereas oxidant release may not be (see later).
Can the Innate Immune Response to Influenza Virus Be Harmful?
There are now several animal model systems in which robust inflammatory responses to influenza impair survival.[56,120,125] In these models, there is sometimes dissociation between viral titers and inflammatory responses. Notably, most of these murine studies were carried out with the A/PR/8 strain, which bypasses lectin-mediated host defense. As previously noted, there is evidence for massive inflammatory response to the avian influenza and 1918 strains.[110,122,123, 126] H5N1 infection of humans and mice causes a similarly severe infection, characterized by high incidence of viral pneumonia and even dissemination beyond the lung, accompanied by massive inflammation.[127,128] These findings raise the question of whether inhibition of some aspects of the inflammatory response could have a beneficial effect in severe cases of influenza. Of particular note in this regard are reactive nitrogen intermediate (RNI) and ROS responses, as well as responses mediated by TLR3 and TNF.
RNI Response
There is an abundant generation of RNI at day 3-6 after A/PR/8 infection in mice.[129-133] NO metabolites can be detected in whole-lung homogenates and also localized to areas of intense viral replication in bronchi of infected mice. NOS2 mediates inducible RNI generation during inflammation and NOS2 -/- mice show improved survival accompanied by reduced pneumonitis in response to influenza. Viral replication is also reduced in this model at day 6 postinfection (although not at early time points). The mechanism for enhanced clearance of virus in the NOS2 -/- mice was found to involve increased production of IFN-γ and elevation of virus-specific IgG2a at days 7 and 9 after infection (correlating with the time of increased viral clearance). Overall, this model appears to reflect a detrimental effect of RNI generation on the immunoglobulin arm of the adaptive response, perhaps through suppressive effects of RNI on CD4 cell recruitment to the lung. RNI do not limit influenza viral replication in tissue culture either.[134,135] Furthermore, ROS and RNIs promote viral mutagenesis and perhaps aid in the evolution of new viral variants.[134,135] It should be noted that there are significant differences in NO metabolism between mice and men, so that one must use caution in direct application of these results to humans.
ROS Response
Reactive oxygen species responses are interwoven with RNI generation in vivo, since superoxide can react with NO to form additional reactive intermediates. The major source of ROS in vivo is the NADPH oxidase of neutrophils and macrophages, although other sources are possible (e.g., xanthine oxidase).[136] Similar to RNI, ROS are mutagenic, potentially damaging to tissues and blockade of ROS generation has been shown to attenuate severity of pneumonitis caused by influenza virus in mice.[137] A mouse model of chronic granulomatous disease has been developed through the deletion of a protein component of the NADPH oxidase. These mice (Cybb tm1 mice) are unable to generate superoxide through the NADPH pathway and have reduced ability to control infections with several strains of bacteria and aspergillus;[119] however, they have reduced influenza viral titers after infection. This surprising result must be evaluated further but it suggests that ROS may not only be harmful due to associated inflammation but may also promote viral replication (or counteract other antiviral mechanisms).
Toll-like Receptor 3
As noted, TLR3 on respiratory epithelial cells plays a role in recognition of influenza viral RNA and triggering of neutrophil recruitment and other inflammatory responses.[54,56] A surprising discovery was that the course of influenza virus infection was actually ameliorated in TLR3 -/- mice,[56] with reduced inflammatory response and improved survival, despite an increase in viral titers. A recent study by Imai et al. demonstrated that H5N1 influenza virus triggers TLR4 activation, leading to acute lung injury.[138] TLR4 activation in these studies was mediated by oxidized phospholipids and was suppressed in mice lacking a protein component of the NADPH oxidase. A similar pathway was activated by several other infectious agents and acid aspiration that also cause acute lung injury. Hence, TLR4 may be an important mediator of oxidant-induced lung injury during severe influenza.
Cytotoxic T Cells and the Role of TNF-α
Cytotoxic T cells directed against the viral HA have been found to contribute to lung injury, principally through the release of TNF-α.[139] The production of TNF-α in this model was found to contribute little to antiviral defense, leading to the conclusion that inhibition of TNF-α production might be of benefit in some cases of severe influenza virus infection. Other cytokines (e.g., IL-6) have also been linked to the severity of influenza symptoms.
Alteration of Innate Defense Caused by Influenza Virus Infection
Influenza virus infection is well known to predispose an individual to asthma exacerbation and bacterial infections. These complications can occur concurrently with influenza or in a delayed manner, days to weeks after the peak of influenza illness.
Bacterial Superinfection
Bacterial pneumonia is a major cause of morbidity and mortality during, or shortly after, influenza epidemics and is a more common complication than viral pneumonia. Influenza virus also predisposes an individual to other infections that are introduced through the respiratory tract, such as bacterial meningitis and otitis media. These findings suggest that influenza infection causes alterations in respiratory immune responses during and after infection. The ability of influenza virus to predispose an individual to bacterial superinfection has been confirmed in animal models and there is mounting evidence that influenza alters innate immunity both early and late (e.g., up to 6 months) after infection.[82,140-143]
Influenza virus alters neutrophil recruitment to the lung and impairs neutrophil antibacterial functions in a broad-based manner, and these alterations correlate with increased susceptibility to bacterial pneumonia or otitis media. In vitro human neutrophil deactivation by influenza virus results from binding of the viral HA to sialylated cell-surface receptors.[144] Preincubation of the virus with anti-HA antibodies or collectins protects neutrophils against this deactivation.[145] The degree of neutrophil dysfunction is also greater in SP-D -/- mice infected with influenza.[146] Influenza causes a slight acceleration of apoptosis of neutrophils, although this process is greatly accelerated when neutrophils are simultaneously exposed to influenza and bacteria.[147] Hence, neutrophil dysfunction may account in part for concurrent bacterial and influenza infection. Influenza virus also causes lytic infection of the airway, which temporarily impairs clearance of bacteria and promotes bacterial adhesion, in part through the action of viral NA.[142]
Of great interest are recent findings that show influenza infection causes more sustained alterations in respiratory responses even after the virus has been cleared. Didierlaurent et al. recently demonstrated that influenza virus infection of mice results in a sustained reduction in TLR2, TLR4 and TLR5 signaling in alveolar macrophages, resulting in reduced cytokine release and reduced neutrophil recruitment.[148] These effects correlate with increased susceptibility to bacterial infection up to 6 weeks after influenza infection. Excessive production of IL-10 weeks after influenza infection has also been linked to delayed bacterial superinfection.[83] Another recent finding of great interest is that the PB1-F2 protein of the 1918 pandemic virus strain contributes to bacterial superinfection, which was a prominent feature of that pandemic.[149] Further research is clearly needed to determine the mechanisms of these effects; however, these fascinating findings strongly suggest that sustained alterations to innate immunity account for the long-standing epidemiological observation of waves of bacterial infection following influenza epidemics. A plausible hypothesis is that the suppression of innate response after influenza viral infection is a homeostatic response to prevent excessive inflammation in the lung environment.
Skewing of Immune Responses to Later Antigen Exposure: The Case of Asthma
Influenza infections can act as a trigger to asthma exacerbation and vaccination of children against influenza reduces asthma attacks.[150,151] Murine models indicate that this phenomenon may be mediated by similar mechanisms to those linked to bacterial superinfection. Influenza infection induces acute type I immune response;[152] however, 1 month after influenza infection, type II-like hypersensitivity responses are strongly potentiated.[152] This effect has been found to relate to a sustained alteration in the polarity of DC responses (i.e., favoring a type II response). Influenza infection can also impair development of tolerance to inhaled antigens in mouse models of respiratory hypersensitivity.[153] SP-D counteracts hypersensitivity lung reactions, which may represent a potential advantage of using recombinant SP-D in therapeutic settings (see later).[154,155]
Future Perspectives
Integrated Picture of Innate and Adaptive Host Defense Against Influenza
It is now clear that the separation between innate and adaptive responses is artificial and that these responses are highly integrated (e.g., see Wright for excellent review of the immunomodulatory effect of SPs;[36]). This is a major topic and we will only highlight some recent findings of interest and point to some valuable reviews.
Innate Mediators Form a Bridge to and Promote Adaptive Immune Responses. At many points in the above discussion, it was noted that innate mediators inform the adaptive response. We cannot do justice to this broad and important topic within this review, but will highlight some important examples of the crosstalk between innate and adaptive immunity. An obvious example of this is the cDC, which forms a direct bridge to the adaptive response by migrating to lymph nodes after viral infection where they present antigen to trafficking T cells leading to clonal expansion. pDCs also direct adaptive responses through type 1 interferon production that promotes CD8+ T-cell activity[156] and immunologic memory.[157] In addition, type 1 interferons directly stimulate B cells soon after influenza viral infection, promoting antiviral antibody responses.[158] An interesting related finding was that either the RIG-I or the TLR7 pathway was sufficient to allow survival from acute influenza viral infection in mice, but the TLR7-MyD88 pathway (used by pDCs) was necessary for development of a protective antibody response.[61] TLR signaling in DCs also potently promotes adaptive response generation[159] and TLR ligands can also directly activate B cells.[160] As noted, C3 and C5 promote T-cell recruitment and responsiveness during influenza virus infection. Even neutrophils may play a role in bridging the innate and adaptive responses by specifically promoting subsequent mononuclear cell infiltration or actually modifying the character of the adaptive response.[161] Defensins also have a variety of effects that could result in modulation of adaptive responses, including chemotactic effects on T cells and DCs.[46] TNF and inducible NOS have been found to promote class switching in IgA generation.[162] It should also be noted that innate mediators can limit the intensity of the adaptive response. NK cells can downregulate CD8+ cell-mediated cytotoxicity[163] and SP-A and SP-D can downregulate T-lymphocyte activation.[36] These effects may help to prevent immune-mediated injury.
Interactions Among Innate Response Components. There is mounting evidence that innate response mediators and signaling pathways are characterized by redundancy and complex interactions and that our current models, which focus only on single components of the response, are inadequate. This is a major topic and one that has not been extensively explored so we will only try to provide some illustrative examples from our work. Collectins have complex interactions with macrophages and neutrophils. Collectins also directly bind to TLRs and CD14 and modify TLR-mediated recognition of some pathogens.[11] Salivary and lung gp-340 bind to SP-D and modulate its activity with respect to influenza virus in vitro.[4,9] Of interest, there are differences in the interaction of salivary gp-340 from different donors with SP-D, as well as differences in interactions of salivary versus lung gp-340 with SP-D. We have noted that α-defensins bind to SP-D and modulate its activity.[49] These examples suggest that our understanding of complex interactions that occur between innate defense mediators is limited and that this area is worthy of more study, especially if some innate inhibitors are to be tested as therapeutics. Of interest, the commonly used NA inhibitor oseltamivir may mediate some of its effects by potentiation activity of innate inhibitors such as mucins and SP-A.[9] One clear lesson of microarray and proteomic studies of the innate response from influenza is that the response is very broad based, involving various families of genes, and that our understanding of the contribution of many specific gene products and the interactions of different gene products is in its infancy.[115]
Expert Commentary and Five-year View
Harnessing Knowledge of Innate Defense to Design Therapies for Influenza
One of the important questions related to influenza is why some subjects suffer severe morbidity or mortality from infection while others have relatively milder, self-limited infection. One possible result of increased understanding of the innate response will be identification of individuals who may be at greater risk of severe influenza. There is already evidence that polymorphisms of TLRs modulate responses to endotoxin or other bacterial infections. It is likely that polymorphisms of the TLRs involved in viral infection affect responses to influenza. There are also polymorphisms of collectins (including MBL, SP-A and SP-D) that have been linked to increased risk of various infections. Similar findings may be obtained with respect to genetic variations in defensins. As already noted, various groups of subjects have acquired deficiencies of SP-D (CF sufferers, COPD patients and smokers) and these groups are more susceptible to influenza. One outcome of discovering susceptibility factors to influenza might be the identification of people most in need of treatment or prophylaxis or people who could most benefit from replacement of particular innate immune mediators (e.g., collectins). See Box 1 for a summary of potential therapuetic approaches.
Box 1. Therapeutic Implications of Innate Immune Research
Recombinant collectins
|
CRD = Carbohydrate- recognition domain; NO = Nitric oxide; NS = Non-structural protein; pDC = Plasmacytoid dendritic cell; RNI = Reactive nitrogen intermediate; ROS = Reactive oxygen species; SP = Surfactant protein; TLR = Toll-like receptor.
Therapeutic Approaches
Recombinant Collectins. SP-D and SP-A have the advantages of being both anti-inflammatory and antiviral, having broad-spectrum activity against bacteria and other pathogens and having other potentially salutary effects on lung function in deficient individuals. Recombinant SP-D has activity against appropriately glycosylated influenza viral strains and might be particularly useful in subjects who lack or have reduced SP-D. Recombinant NCRD trimers mediate many of the beneficial effects of SP-D when administered intranasally in mice, including promotion of clearance of apoptotic cells, downregulation of NO response, reduction of allergic response to fungi and improvement of the outcome of RSV infection.[154,164-168] There are also possible advantages in NCRD preparations, since they lack the collagen domain that is thought to bind to proinflammatory receptors. NCRDs can be produced in Escherichia coli, whereas full-length SP-D must be generated in mammalian cell cultures. Unfortunately, wild-type NCRD of SP-D has greatly diminished inhibitory activity for influenza viruses (including strains susceptible to full-length SP-D).[169] However, we have found that relatively limited modifications of the SP-D NCRD confer antiviral activity (Figure 1), including insertion of the three amino acid sequences, RAK or AAA where indicated[169] or substitutions for R343 (Hartshorn KL, Crouch E, Unpublished Data). Of interest, it is also possible to increase activity of NCRD by retaining the N-terminus in the absence of a collagen domain (resulting in multimers that retain viral neutralization activity in the absence of a collagen domain).[17] In addition, crosslinking of the NCRD with antibodies that do not attach to, or interfere with, the binding surface of the CRD potentiates antiviral activity.[170] Coupling of the SP-D NCRD to a Fab1 fragment of an antibody directed against the IgA Fc receptor results in a molecule that can greatly potentiate phagocytosis of Candida, bacteria and influenza virus by neutrophils.[171] Porcine SP-D can be used as a template in the design of collectin-based antivirals because of its distinctive properties.[38] A limitation of SP-D as an antiviral is its possible lack of activity against pandemic strains. Porcine SP-D or SP-A, or pentraxins, may, however, have increased activity against such strains.
Defensins. Defensins can be synthesized in large quantities and they also have broad-spectrum activity, including inhibition of many types of bacteria, fungi and enveloped viruses. They can act as opsonins and also have a variety of interactions with adaptive immune effector cells.[50] Structure-function studies with synthesized defensins have identified changes that increase antiviral activity, sometimes through alteration of single amino acids. Studies are underway in our laboratory to evaluate antiviral activity and immune interactions of panels of synthetic defensins. Potential limitations of defensins as therapeutics are the possible induction of inflammation[172] or inhibition of functions of SP-D.[49]
TLR Ligands. Several papers have demonstrated that upregulation or stimulation of innate RNA-recognition pathways can improve outcome of influenza virus infection. For instance, stimulation of TLR7 with synthetic agonists reduced the severity of influenza in rats and mice.[173,174]
Increase Type 1 Interferon Production or DC Activation. Stimulation of type 1 interferon responses through manipulation of TLRs appears to be a promising approach because this could lead to localized production of interferon at sites of infection (i.e., by lowering the threshold for production in infected cells). This could be accomplished through TLR activation, counteracting the effects of NS1 or other measures, such as those that could promote DC activation.[72,92] Viral strains lacking the NS1 or with mutated versions of NS1 have been produced through reverse genetics and these have an attenuated phenotype that could make them useful as vaccines.
Blockade of Inflammatory Response Mediators: The Controversy
As outlined earlier, some specific aspects of the innate response to influenza may be harmful and may provide therapeutic targets, including oxidant generation, NF-κB activation and TNF-α generation. As an example, blockade of NF-κB by aspirin led to a reduction of viral growth in vitro and in vivo in mice, retention of viral RNP in the nucleus of infected cells in vitro and improved survival from lethal influenza in mice.[175] As noted, inhibition of oxidant generation or TLR3 activitation was also protective in mice. By contrast, prevention of IL-1 activity was not protective. IL-1 is released in the early phases of influenza infection and has potent proinflammatory effects, including regulation of CXC chemokines and promotion of early neutrophil influx.[82] The course of A/PR/8 infection in IL-1R -/- mice was characterized by reduced inflammatory response, including reduced neutrophil infiltration, but viral titers were mildly elevated in these mice and mortality was greatly increased. As noted, blockade of macrophage responses in pigs was harmful.[108] Hence, it will be necessary to dissect out which specific innate responses are protective versus harmful.
A further challenge is to extrapolate murine findings to other models that more closely resemble human infection or to human infection itself, in order to clarify which aspects of the innate response can be safely inhibited without compromising antiviral activity. The macaque model has been very valuable in this regard and the ferret model may be useful in the future. One can draw limited conclusions from clinical studies in humans thus far. For instance, a recent study showed that infants dying of RSV or influenza in the USA did not show evidence of excessive immune pathology but rather there was suggestion of blunted immune responses in these cases.[176] Furthermore, many of the subject groups most prone to complications of influenza have diminished immune competence, including the elderly (diminished T-cell responsiveness and TLR activity),[177-179] subjects with HIV or those immune compromised by bone marrow transplantation, pregnant women and infants. Subjects with diabetes mellitus, CF or COPD may also be more susceptible because of limitations in innate immunity (e.g., diminished SP-D levels or function).
Pandemic Viruses: A Special Case? An important question is whether infection with the 1918 or H5N1 viruses (or other prior or future pandemic strains) are special cases in which immunopathology plays a greater role.
In vitro infection of macrophages with highly pathogenic H5N1 strains results in high cytokine release.[81] This finding, coupled with the massive parenchymal lung pathology found in human cases, suggests that H5N1 infection may be more severe, in part because of dysregulated inflammatory response. On the other hand, there is evidence that H5N1 strains may be particularly effective at counteracting or evading immune responses. NS1 of lethal H5N1 is particularly effective at inhibiting antiviral cytokine responses.[180] H5N1 is also particularly effective at replicating in cDCs and depleting these cells, which may result in crippling of the adaptive immune response.[98] As noted, H5N1 strains are not inhibited by SP-D. A study of human subjects with fatal H5N1 infection revealed increased viral loads in the pharynx compared with standard human influenza infection, and also noted detection of virus in blood and the rectum (not seen in human influenza).[128] High viral loads correlated with an intense cytokine response, inflammation and depletion of T lymphocytes. These results suggest that the inflammatory response may in fact be secondary to high viral replication and dissemination of the virus to the lungs and other sites, rather than an autonomous response that should be targeted for treatment in its own right. Corticosteroids have been tested in H5N1-infected subjects but they have not been shown to be beneficial and their use is not recommended.[127] Furthermore, a recent study showed that mice lacking key proinflammatory cytokines or treated with corticosteroids did not have reduced mortality with H5N1 infection.[181] In terms of the 1918 strain, the overall intensity of inflammatory responses seems to correlate with intensity of viral replication, rather than be dissociated from it (see discussion above). Hence, even in these instances, antiviral therapy and vaccine development would appear to be the most compelling goals until more targeted anti-inflammatory therapies can be developed.
Sidebar: Key Issues
- The innate immune response is much more complex than previously appreciated and is characterized by extensive fine tuning and remarkable specificity, despite the fact that this branch of immunity is hardwired in the human genome.
- Influenza virus infection is a prime example of the success of innate immunity, given the daunting task of containing a constantly changing virus while minimizing inflammatory injury in the delicate lung environment.
- It is increasingly clear that there are both positive and negative regulators of the innate response (e.g., see recent findings related to type 1 interferon response). It is also clear that the innate response plays a key role in directing the adaptive response and that there is a high degree of interplay between innate and adaptive immunity.
- The abundance of information regarding the innate response to influenza promises to clarify certain key issues, including why certain subjects are more prone to complications and why certain viral strains are much more pathogenic (either through eliciting more intense inflammation or increased effectiveness at counteracting innate responses via NS1 or PB1-F2).
- It is remarkable that some innate mediators (e.g., Toll-like receptors, collectins and defensins) have broad-spectrum antimicrobial activity while retaining considerable specificity.
- These findings can be exploited to develop improved therapies or vaccine strategies for influenza. This approach can be thought of as using the 'wisdom of the body' to guide development of therapies for the future.
References
- Morens DM. Influenza-related mortality. JAMA 289, 227-229 (2003).
- Garcia-Sastre A, Whitley RJ. Lessons learned from reconstructing the 1918 influenza pandemic. J. Infect. Dis. 194(Suppl. 2), S127-S132 (2006).
- Finberg RW, Wang JP, Kurt-Jones EA. Toll like receptors and viruses. Rev. Med. Virol. 17(1), 35-43 (2007).
- Hartshorn KL, Ligtenberg A, White MR et al. Salivary agglutinin and lung scavenger receptor cysteine-rich glycoprotein 340 have broad anti-influenza activities and interactions with surfactant protein D that vary according to donor source and sialylation. Biochemistry 393, 545-553 (2006).
- Hartshorn KL, White MR, Mogues T et al. Lung and salivary scavenger receptor glycoprotein- 340 contribute to the host defense against influenza A viruses. Am. J. Physiol. Lung Cell. Mol. Physiol. 285(5), L1066-L1076 (2003).
- Brauer L, Kindler C, Jager K et al. Detection of surfactant proteins A and D in human tear fluid and the human lacrimal system. Invest. Ophthalmol. Vis. Sci. 48(9), 3945-3953 (2007).
- Belser JA, Lu X, Maines TR et al. Pathogenesis of avian influenza (H7) virus infection in mice and ferrets: enhanced virulence of Eurasian H7N7 viruses isolated from humans. J. Virol. 81(20), 11139-11147 (2007).
- Reading PC, Bozza S, Gilbertson B et al. Antiviral activity of the long chain pentraxin PTX3 against influenza viruses. J. Immunol. 180(5), 3391-3398 (2008).
- White MR, Crouch E, van Eijk M et al. Cooperative anti-influenza activities of respiratory innate immune proteins and neuraminidase inhibitor. Am. J. Physiol. Lung Cell. Mol. Physiol. 288(5), L831-L840 (2005).
- White MR, Crouch E, Vesona J et al. Respiratory innate immune proteins differentially modulate the neutrophil respiratory burst response to influenza A virus. Am. J. Physiol. Lung Cell. Mol. Physiol. 289(4), L606-L616 (2005).
- Kuroki Y, Takahashi M, Nishitani C. Pulmonary collectins in innate immunity of the lung. Cell. Microbiol. 9(8), 1871-1879 (2007).
- Whitsett JA. Surfactant proteins in innate host defense of the lung. Biol. Neonate 88(3), 175-180 (2005).
- Hartshorn KL, Crouch EC, White MR et al. Evidence for a protective role of pulmonary surfactant protein D (SP-D) against influenza A viruses. J. Clin. Invest. 94, 311-319 (1994).
- Madsen J, Kliem A, Tornoe I et al. Localization of lung surfactant protein D on mucosal surfaces in human tissues. J. Immunol. 164, 5866-5870 (2000).
- LeVine AM, Whitsett JA, Hartshorn KL, Crouch EC, Korfhagen TR. Surfactant protein D enhances clearance of influenza A virus from the lung in vivo. J. Immunol. 167(10), 5868-5873 (2001).
- Hawgood S, Brown C, Edmondson J et al. Pulmonary collectins modulate strain-specific influenza A virus infection and host responses. J. Virol. 78(16), 8565-8572 (2004).
- Kingma PS, Zhang L, Ikegami M et al. Correction of pulmonary abnormalities in Sftpd-/- mice requires the collagenous domain of surfactant protein D. J. Biol. Chem. 281(34), 24496-24505 (2006).
- Zhang L, Hartshorn K, Crouch E, Ikegami M, Whitsett J. Complementation of pulmonary abnormalities in SP-D -/- mice with an SP-D/conglutinin fusion protein. J. Biol. Chem. 277, 22453-22459 (2002).
- Reading P, Morey L, Crouch E, Anders E. Collectin-mediated antiviral host defense of the lung: evidence from influenza virus infection of mice. J. Virol. 71, 8204-8212 (1997).
- Hartley C, Reading P, Ward A, Anders E. Changes in hemagglutinin molecule of influenza type A (H3N2) virus associated with increased virulence in mice. Arch. Virol. 142, 75-88 (1997).
- Hartshorn KL, Sastry K, White MR et al. Human mannose-binding protein functions as an opsonin for influenza A viruses. J. Clin. Invest. 91, 1414-1420 (1993).
- Vigerust DJ, Ulett KB, Boyd KL et al. N-linked glycosylation attenuates H3N2 influenza viruses. J. Virol. 81(16), 8593-8600 (2007).
- Stevens J, Corper AL, Basler CF et al. Structure of the uncleaved human H1 hemagglutinin from the extinct 1918 influenza virus. Science 303(5665), 1866-1870 (2004).
- Skehel JJ, Wiley DC. Influenza haemagglutinin. Vaccine 20(Suppl. 2), S51-S54 (2002).
- Mitnaul LJ, Matrosovich MN, Castrucci MR et al. Balanced hemagglutinin and neuraminidase activities are critical for efficient replication of influenza a virus. J. Virol. 74(13), 6015-6020 (2000).
- Tsuchiya E, Sugawara K, Hongo S et al. Effect of addition of new oligosaccharide chains to the globular head of influenza A/H2N2 virus haemagglutinin on the intracellular transport and biological activities of the molecule. J. Gen. Virol. 83(Pt 5), 1137-1146 (2002).
- Honda Y, Takahashi H, Kuroki Y, Akino T, Abe S. Decreased contents of surfactant proteins A and D in BAL fluids of healthy smokers. Chest 109, 1006-1009 (1996).
- Postle A, Mander A, Reid K et al. Deficient Hydrophilic surfactant proteins A and D with normal surfactant phospholipid molecular species in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 20, 90-98 (1999).
- Sims MW, Tal-Singer RM, Kierstein S et al. Chronic obstructive pulmonary disease and inhaled steroids alter surfactant protein D (SP-D) levels: a cross-sectional study. Respir. Res. 9, 13 (2008).
- White MR, Tecle T, Crouch EC, Hartshorn KL. Impact of neutrophils on antiviral activity of human bronchoalveolar lavage fluid. Am. J. Physiol. Lung Cell. Mol. Physiol. 293(5), L1293-L1299 (2007).
- Reading P, Allison J, Crouch E, Anders E. Increased susceptibility of diabetic mice to influenza virus infection: compromise of collectin-mediated host defense of the lung by glucose. J. Virol. 72, 6884-6887 (1998).
- Hartshorn KL, White MR, Tecle T et al. Reduced influenza viral neutralizing activity of natural human trimers of surfactant protein D. Respir. Res. 8, 9 (2007).
- LahtI M, Lofgren J, Martilla T et al. Surfactant protein D gene polymorphism associated with severe respiratory syncytial virus infection. Pediatr. Res. 51(6), 696-699 (2002).
- Floros J, Lin H, Garcia A et al. Surfactant protein marker alleles identify a subgroup of tuberculosis patients in a Mexican population. J. Infect. Dis. 182, 1473-1478 (2000).
- Koch A, Melbye M, Sorensen P et al. Acute respiratory tract infections and mannose-binding lectin insufficiency during early childhood. JAMA 285, 1316-1321 (2001).
- Wright JR. Immunoregulatory functions of surfactant proteins. Nat. Rev. Immunol. 5(1), 58-68 (2005).
- Hartshorn KL, White MR, Shepherd V et al. Mechanisms of anti-influenza activity of surfactant proteins A and D: comparison with serum collectins. Am. J. Physiol. 273(6 Pt 1), L1156-L1166 (1997).
- van Eijk M, White MR, Batenburg JJ et al. Interactions of influenza A virus with sialic acids present on porcine surfactant protein D. Am. J. Respir. Cell Mol. Biol. 30(6), 871-879 (2004).
- Baumgarth N, Herman OC, Jager GC et al. B-1 and B-2 cell-derived immunoglobulin M antibodies are nonredundant components of the protective response to influenza virus infection. J. Exp. Med. 192(2), 271-280 (2000).
- Jayasekera JP, Moseman EA, Carroll MC. Natural antibody and complement mediate neutralization of influenza virus in the absence of prior immunity. J. Virol. 81(7), 3487-3494 (2007).
- Watford W, Chio A, Wright J. Complement-mediated host defense in the lung. Am. J. Physiol. 279, L790-L798 (2000).
- Ezekowitz RA. Role of the mannose-binding lectin in innate immunity. J. Infect. Dis. 187(Suppl. 2), S335-S339 (2003).
- Anders EM, Hartley CA, Reading PC, Ezekowitz RAB. Complement-dependen t neutralization of influenza virus by a serum mannose-binding lectin. J. Gener. Virol. 75, 615-622 (1994).
- Kim AH, Dimitriou ID, Holland MC et al. Complement C5a receptor is essential for the optimal generation of antiviral CD8+ T cell responses. J. Immunol. 173(4), 2524-2529 (2004).
- Kopf M, Abel B, Gallimore A, Carroll M, Bachmann MF. Complement component C3 promotes T-cell priming and lung migration to control acute influenza virus infection. Nat. Med. 8(4), 373-378 (2002).
- Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat. Rev. 3, 710-720 (2003).
- Salvatore M, Garcia-Sastre A, Ruchala P et al. α-defensin inhibits influenza virus replication by cell-mediated mechanism(s) . J. Infect. Dis. 196(6), 835-843 (2007).
- Daly NL, Chen YK, Rosengren KJ et al. Retrocyclin- 2: structural analysis of a potent anti-HIV t-defensin. Biochemistry 46(35), 9920-9928 (2007).
- Hartshorn KL, White MR, Tecle T, Holmskov U, Crouch EC. Innate defense against influenza A virus: activity of human neutrophil defensins and interactions of defensins with surfactant protein D. J. Immunol. 176(11), 6962-6972 (2006).
- Tecle T, White MR, Gantz D, Crouch EC, Hartshorn KL. Human neutrophil defensins increase neutrophil uptake of influenza A virus and bacteria and modify virus-induced respiratory burst responses. J. Immunol. 178(12), 8046-8052 (2007).
- Seth RB, Sun L, Chen ZJ. Antiviral innate immunity pathways. Cell Res. 16(2), 141-147 (2006).
- Diebold S, Kaisho T, Hemmi H, Akira S, Sousa CE. Innate antiviral responses by means of TLR7-mediated recognition of single stranded RNA. Science 303, 1529-1531 (2004).
- Wang JP, Liu P, Latz E et al. Flavivirus activation of plasmacytoid dendritic cells delineates key elements of TLR7 signaling beyond endosomal recognition. J. Immunol. 177(10), 7114-7121 (2006).
- Le Goffic R, Pothlichet J, Vitour D et al. Cutting edge: influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J. Immunol. 178(6), 3368-3372 (2007).
- Guillot L, Le Goffic R, Bloch S et al. Involvement of Toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J. Biol. Chem. 280(7), 5571-5580 (2005).
- Le Goffic R, Balloy V, Lagranderie M et al. Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog. 2(6), e53 (2006).
- Pichlmair A, Schulz O, Tan CP et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5´-phosphates. Science 314(5801), 997-1001 (2006).
• This, and an additional article in the same issue of Science, demonstrates how retinoid-inducible gene (RIG)-1 can specifically recognize RNA virus infection in host cells. - Loo YM, Fornek J, Crochet N et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 82(1), 335-345 (2008).
- Kato H, Takeuchi O, Sato S et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441(7089), 101-105 (2006).
- Sun Q, Sun L, Liu HH et al. The specific and essential role of MAVS in antiviral innate immune responses. Immunity 24(5), 633-642 (2006).
- Koyama S, Ishii KJ, Kumar H et al. Differential role of TLR- and RLR-signaling in the immune responses to influenza A virus infection and vaccination. J. Immunol. 179(7), 4711-4720 (2007).
- Lund JM, Alexopoulou L, Sato A et al. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl Acad. Sci. USA 101(15), 5598-5603 (2004).
- Wurzer WJ, Ehrhardt C, Pleschka S et al. NF-κB-dependent induction of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and Fas/FasL is crucial for efficient influenza virus propagation. J. Biol. Chem. 279(30), 30931-30937 (2004).
- Wurzer WJ, Planz O, Ehrhardt C et al. Caspase 3 activation is essential for efficient influenza virus propagation. EMBO J. 22(11), 2717-2728 (2003).
- Bernasconi D, Amici C, La Frazia S, Ianaro A, Santoro MG. The IκB kinase is a key factor in triggering influenza A virus-induced inflammatory cytokine production in airway epithelial cells. J. Biol. Chem. 280(25), 24127-24134 (2005).
- Wei L, Sandbulte MR, Thomas PG et al. NFκB negatively regulates interferon-induced gene expression and anti-influenza activity. J. Biol. Chem. 281(17), 11678-11684 (2006).
- Pothlichet J, Chignard M, Si-Tahar M. Cutting edge: innate immune response triggered by influenza A virus is negatively regulated by SOCS1 and SOCS3 through a RIG-I/IFNAR1- dependent pathway. J. Immunol. 180(4), 2034-2038 (2008).
• Provides some insight into negative regulatory feedback mechanisms for innate immune responses during influenza viral infection. - Marjuki H, Alam MI, Ehrhardt C et al. Membrane accumulation of influenza A virus hemagglutinin triggers nuclear export of the viral genome via protein kinase Ca-mediated activation of ERK signaling. J. Biol. Chem. 281(24), 16707-16715 (2006).
- Hartshorn KL, Wright J, Collamer M, White MR, Tauber AI. Human neutrophil stimulation by influenza virus: relationship of cytoplasmic pH changes to cell activation. Am. J. Physiol. 258, C1070-C1076 (1990).
- Hartshorn KL, Collamer M, White MR, Schwartz JH, Tauber AI. Characterization of influenza A virus activation of the human neutrophil. Blood 75(1), 218-226 (1990).
- Lee DC, Cheung CY, Law AH et al. p38 mitogen-activated protein kinase-dependent hyperinduction of tumor necrosis factor a expression in response to avian influenza virus H5N1. J. Virol. 79(16), 10147-10154 (2005).
- Garcia-Sastre A. Antiviral response in pandemic influenza viruses. Emerg. Infect. Dis. 12(1), 44-47 (2006).
- Garcia-Sastre A, Durbin R, Zheng H et al. The role of interferon in influenza virus tissue tropism. J. Virol. 72, 8550-8558 (1998).
- Price GE, Gaszewska-Mastarlar z A, Moskophidis D. The role of α/β and g interferons in development of immunity to influenza A virus in mice. J. Virol. 74(9), 3996-4003 (2000).
- Wang X, Ming L, Zheng H et al. Influenza A virus NS1 protein prevents activation of NFκB and induction of α/β interferon. J. Virol. 74, 11566-11573 (2000).
- Geiss G, Salvatore M, Tumpey T et al. Cellular transcription profiling in influenza A virus-infected lung epithelial cells: the role of the nonstructural NS1 protein in the evasion of the host innate defense and its potential contribution to pandemic influenza. Proc. Natl Acad. Sci. USA 99, 10736-10741 (2002).
- Jewell NA, Vaghefi N, Mertz SE et al. Differential type I interferon induction by respiratory syncytial virus and influenza A virus in vivo. J. Virol. 81(18), 9790-9800 (2007).
- Hayden F, Fritz R, Lobo M et al. Local and systemic cytokine response during experimental human influenza A virus infection. J. Clin. Invest. 101, 643-649 (1998).
- Lee N, Wong CK, Chan PK et al. Hypercytokinemia and hyperactivation of phospho-p38 mitogen-activated protein kinase in severe human influenza A virus infection. Clin. Infect. Dis. 45(6), 723-731 (2007).
- Denton AE, Doherty PC, Turner SJ, La Gruta NL. IL-18, but not IL-12, is required for optimal cytokine production by influenza virus-specific CD8+ T cells. Eur. J. Immunol. 37(2), 368-375 (2007).
- Szretter KJ, Gangappa S, Lu X et al. Role of host cytokine responses in the pathogenesis of avian H5N1 influenza viruses in mice. J. Virol. 81(6), 2736-2744 (2007).
- Schmitz N, Kurrer M, Bachmann MF, Kopf M. Interleukin- 1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J. Virol. 79(10), 6441-6448 (2005).
- Van Der Sluijs KF, Van Elden LJ, Nijhuis M et al. IL-10 is an important mediator of the enhanced susceptibility to pneumococcal pneumonia after influenza infection. J. Immunol. 172(12), 7603-7609 (2004).
- Zeng H, Goldsmith C, Thawatsupha P et al. Highly pathogenic avian influenza H5N1 viruses elicit an attenuated type I interferon response in polarized human bronchial epithelial cells. J. Virol. 81(22), 12439-12449 (2007).
- Chan MC, Cheung CY, Chui WH et al. Proinflammatory cytokine responses induced by influenza A (H5N1) viruses in primary human alveolar and bronchial epithelial cells. Respir. Res. 6, 135 (2005).
- Matrosovich MN, Matrosovich TY, Gray T, Roberts NA, Klenk H. Human and avian influenza viruses target different cell types in cultures of human airway epithelium. Proc. Natl Acad. Sci. USA 101(13), 4620-4624 (2004).
- van Riel D, Munster VJ, de Wit E et al. H5N1 Virus attachment to lower respiratory tract. Science 312(5772), 399 (2006).
- Kido H, Yokogoshi Y, Sakai K, Tashiro M, Kishino Y. Isolation and characterization of a novel trypsin-like protease found in rat bronchiolar epithelial clara cells. J. Biol. Chem. 267, 13573-13579 (1992).
- Hashimoto Y, Moki T, Takizawa T, Shiratsuchi A, Nakanishi Y. Evidence for phagocytosis of influenza virus-infected, apoptotic cells by neutrophils and macrophages in mice. J. Immunol. 178(4), 2448-2457 (2007).
- Arndt U, Wennemuth G, Barth P et al. Release of macrophage migration inhibitory factor and CXCL8/interleukin- 8 from lung epithelial cells rendered necrotic by influenza A virus infection. J. Virol. 76(18), 9298-9306 (2002).
- Meyerholz DK, Kawashima K, Gallup JM, Grubor B, Ackermann MR. Expression of select immune genes (surfactant proteins A and D, sheep β defensin 1, and Toll-like receptor 4) by respiratory epithelia is developmentally regulated in the preterm neonatal lamb. Dev. Comp. Immunol. 30(11), 1060-1069 (2006).
- Grayson MH, Holtzman MJ. Emerging role of dendritic cells in respiratory viral infection. J. Mol. Med. 85(10), 1057-1068 (2007).
• Excellent review on the role of dendritic cells in respiratory virus infection. - Lopez CB, Moltedo B, Alexopoulou L et al. TLR-independent induction of dendritic cell maturation and adaptive immunity by negative-strand RNA viruses. J. Immunol. 173(11), 6882-6889 (2004).
- Colina R, Costa-Mattioli M, Dowling RJ et al. Translational control of the innate immune response through IRF-7. Nature 452(7185), 323-328 (2008).
- Dai J, Megjugorac NJ, Amrute SB, Fitzgerald-Bocarsly P. Regulation of IFN regulatory factor-7 and IFN-α production by enveloped virus and lipopolysaccharide in human plasmacytoid dendritic cells. J. Immunol. 173(3), 1535-1548 (2004).
- Piqueras B, Connolly J, Freitas H, Palucka AK, Banchereau J. Upon viral exposure, myeloid and plasmacytoid dendritic cells produce 3 waves of distinct chemokines to recruit immune effectors. Blood 107(7), 2613-2618 (2006).
- Draghi M, Pashine A, Sanjanwala B et al. NKp46 and NKG2D recognition of infected dendritic cells is necessary for NK cell activation in the human response to influenza infection. J. Immunol. 178(5), 2688-2698 (2007).
- Thitithanyanont A, Engering A, Ekchariyawat P et al. High susceptibility of human dendritic cells to avian influenza H5N1 virus infection and protection by IFN-α and TLR ligands. J. Immunol. 179(8), 5220-5227 (2007).
- Fernandez-Sesma A, Marukian S, Ebersole BJ et al. Influenza virus evades innate and adaptive immunity via the NS1 protein. J. Virol. 80(13), 6295-6304 (2006).
- Gazit R, Gruda R, Elboim M et al. Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nat. Immunol. 7(5), 517-523 (2006).
- Mandelboim O, Lieberman N, Lev M et al. Recognition of haemagglutinins of virus-infected cells by NKp46 activates lysis by NK cells. Nature 409, 1055-1060 (2001).
- Ho JW, Hershkovitz O, Peiris M et al. H5-type influenza hemagglutinin is functionally recognized by the natural killer activating receptor, NKp44. J. Virol. 82(4), 2028-2032 (2007).
- Owen RE, Yamada E, Thompson CI et al. Alterations in receptor binding properties of recent human influenza H3N2 viruses are associated with reduced natural killer cell lysis of infected cells. J. Virol. 81(20), 11170-11178 (2007).
- Watanabe Y, Hashimoto Y, Shiratsuchi A, Takizawa T, Nakanishi Y. Augmentation of fatality of influenza in mice by inhibition of phagocytosis. Biochem. Biophys. Res. Commun. 337(3), 881-886 (2005).
- van der Sluijs KF, van Elden L, Nijhuis M et al. Toll-like receptor 4 is not involved in host defense against respiratory tract infection with Sendai virus. Immunol. Lett. 89(2-3), 201-206 (2003).
- Cassidy L, Lyles D, Abramson J. Synthesis of viral proteins in polymorphonuclear leukocytes infected with influenza A virus. J. Clin. Microbiol. 26, 1267-1270 (1988).
- Sweet C, Bird RA, Howie AJ et al. Further studies of the reasons for the lack of alveolar infection during influenza in ferrets. Br. J. Exp. Pathol. 66(2), 217-231 (1985).
- Kim HM, Lee YW, Lee KJ et al. Alveolar macrophages are indispensable for controlling influenza viruses in lungs of pigs. J. Virol. 82(9), 4265-4274 (2008).
- Fujisawa H, Tsuru S, Taniguchi M, Zinnaka Y, Nomoto K. Protective Mechanisms against pulmonary infection with influenza virus. I. Relative contribution of polymorphonuclear leukocytes and of alveolar mactrophages to protection during the early phase of intranasal infection. J. Gen. Virol. 68, 425-432 (1987).
- Tumpey TM, Garcia-Sastre A, Taubenberger JK et al. Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J. Virol. 79(23), 14933-14944 (2005).
- Conenello GM, Zamarin D, Perrone LA, Tumpey T, Palese P. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog. 3(10), 1414-1421 (2007).
- Zamarin D, Garcia-Sastre A, Xiao X, Wang R, Palese P. Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog. 1(1), e4 (2005).
- Lin KL, Suzuki Y, Nakano H, Ramsburg E, Gunn MD. CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J. Immunol. 180(4), 2562-2572 (2008).
- Baskin CR, Bielefeldt-Ohmann H, Garcia-Sastre A et al. Functional genomic and serological analysis of the protective immune response resulting from vaccination of macaques with an NS1-truncated influenza virus. J. Virol. 81(21), 11817-11827 (2007).
- Baas T, Baskin CR, Diamond DL et al. Integrated molecular signature of disease: analysis of influenza virus-infected macaques through functional genomics and proteomics. J. Virol. 80(21), 10813-10828 (2006).
•• Tour de force showing the value of the macaque model and high-throughput genomics and proteomics in understanding the immune response to influenza. - Baskin CR, Garcia-Sastre A, Tumpey TM et al. Integration of clinical data, pathology, and cDNA microarrays in influenza virus-infected pigtailed macaques (Macaca nemestrina). J. Virol. 78(19), 10420-10432 (2004).
- Smith H, Sweet C. Lessons for human influenza from pathogenicity studies with ferrets. Rev. Infect. Dis. 10, 56-75 (1988).
- Wareing MD, Shea AL, Inglis CA, Dias PB, Sarawar SR. CXCR2 is required for neutrophil recruitment to the lung during influenza virus infection, but is not essential for viral clearance. Viral Immunol. 20(3), 369-378 (2007).
- Snelgrove RJ, Edwards L, Rae AJ, Hussell T. An absence of reactive oxygen species improves the resolution of lung influenza infection. Eur. J. Immunol. 36(6), 1364-1373 (2006).
- Sakai S, Kawamata H, Mantani N et al. Therapeutic effect of anti-macrophage inflammatory protein 2 antibody on influenza virus-induced pneumonia in mice. J. Virol. 74(5), 2472-2476 (2000).
- Tsuru S, Fujisawa H, Taniguchi M, Zinnaka Y, Nomoto K. Mechanism of protection during the early phase of a generalized viral infection II. Contribution of polymorphonuclear leukocytes to protection against intravenous infection with influenza virus. J. Gen. Virol. 68, 419-424 (1987).
- Kobasa D, Jones SM, Shinya K et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature 445(7125), 319-323 (2007).
- Kobasa D, Takada A, Shinya K et al. Enhanced virulence of influenza A viruses with the haemagglutinin of the 1918 pandemic virus. Nature 431(7009), 703-707 (2004).
• This and other papers by this group demonstrated the high degree of pathogenicity and profound innate immune response triggered by recombinant strains of the 1918 virus. - Hartshorn KL, Daigneault DE, White MR, Tauber AI. Anomalous features of human neutrophil activation by influenza A virus are shared by related viruses and sialic acid-binding lectins. J. Leuk. Biol. 51, 230-236 (1992).
- Dawson TC, Beck MA, Kuziel WA, Henderson F, Maeda N. Contrasting effects of CCR5 and CCR2 deficiency in the pulmonary inflammatory response to influenza A virus. Am. J. Pathol. 156(6), 1951-1959 (2000).
- Tumpey T, Garcia-Sastre A, Taubenberger J, Palese P, Swayne D. Pathogenicity and immunogenicity of influenza viruses with genes from the 1918 pandemic virus. Proc. Natl Acad. Sci. USA 101(9), 3166-3171 (2004).
- Abdel-Ghafar AN, Chotpitayasunondh T, Gao Z et al. Update on avian influenza A (H5N1) virus infection in humans. N. Engl. J. Med. 358(3), 261-273 (2008).
- de Jong MD, Simmons CP, ThanhTT et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat. Med. 12(10), 1203-1207 (2006).
- Jayasekera JP, Vinuesa CG, Karupiah G, King NJ. Enhanced antiviral antibody secretion and attenuated immunopathology during influenza virus infection in nitric oxide synthase-2-deficien t mice. J. Gen. Virol. 87(Pt 11), 3361-3371 (2006).
- Karupiah G, Chen JH, Mahalingam S, Nathan CF, MacMicking JD. Rapid interferon γ-dependent clearance of influenza A virus and protection from consolidating pneumonitis in nitric oxide synthase 2-deficient mice. J. Exp. Med. 188(8), 1541-1546 (1998).
- Zaki MH, Akuta T, Akaike T. Nitric oxide-induced nitrative stress involved in microbial pathogenesis. J. Pharmacol. Sci. 98(2), 117-129 (2005).
- Akaike T, Okamoto S, Sawa T et al. 8-nitroguanosine formation in viral pneumonia and its implication for pathogenesis. Proc. Natl Acad. Sci. USA 100(2), 685-690 (2003).
- Akaike T, Noguchi Y, Ijiri S et al. Pathogenesis of influenza virus-induced pneumonia: involvement of both nitric oxide and oxygen radicals. Proc. Natl Acad. Sci. USA 93, 2448-2453 (1996).
- Yoshitake J, Akaike T, Akuta T et al. Nitric oxide as an endogenous mutagen for Sendai virus without antiviral activity. J. Virol. 78(16), 8709-8719 (2004).
- Akaike T. Role of free radicals in viral pathogenesis and mutation. Rev. Med. Virol. 11(2), 87-101 (2001).
- Akaike T, Ando M, Oda T et al. Dependence on O2- generation by xanthine oxidase of pathogenesis of influenza virus infection in mice. J. Clin. Invest. 85(3), 739-745 (1990).
- Oda T, Akaike T, Hamamoto T et al. Oxygen radicals in influenza-induced pathogenesis and treatment with pyran polymer-conjugated SOD. Science 244(4907), 974-976 (1989).
- Imai Y, Kuba K, Neely GG et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 133(2), 235-249 (2008).
•• Shows the role of Toll-like receptor (TLR)4 in a common pathway of acute lung injury triggered by H5N1, SARS, anthrax and acid aspiration. - Xu L, Yoon H, Zhao MQ et al. Cutting edge: pulmonary immunopathology mediated by antigen-specific expression of TNF-α by antiviral CD8+ T cells. J. Immunol. 173(2), 721-725 (2004).
- Speshock JL, Doyon-Reale N, Rabah R, Neely MN, Roberts PC. Filamentous influenza A virus infection predisposes mice to fatal septicemia following superinfection with Streptococcus pneumoniae serotype 3. Infect. Immun. 75(6), 3102-3111 (2007).
- McNamee LA, Harmsen AG. Both influenza-induced neutrophil dysfunction and neutrophil-independ ent mechanisms contribute to increased susceptibility to a secondary Streptococcus pneumoniae infection. Infect. Immun. 74(12), 6707-6721 (2006).
- McCullers JA, Bartmess KC. Role of neuraminidase in lethal synergism between influenza virus and Streptococcus pneumoniae. J. Infect. Dis. 187, 1000-1009 (2003).
- Abramson JS, Giebink GS, Quie PG. Influenza A virus-induced polymorphonuclear leukocyte dysfunction in the pathogenesis of experimental pneumococcal otitis media. Infect. Immun. 36, 289-296 (1982).
- Hartshorn KL, Liou LS, White MR, Kazhdan MM, Tauber JL. Neutrophil deactivation by influenza A virus: role of hemagglutinin binding to specific sialic acid-bearing cellular proteins. J. Immunol. 154, 3952-3960 (1995).
- Hartshorn K, Reid K, White M et al. Neutrophil deactivation by influenza A viruses: mechanisms of protection after viral opsonization with collectins and hemagglutination- inhibiting antibodies. Blood 87, 3450-3461 (1996).
- LeVine A, Whitsett J, Hartshorn K, Korfhagen T. Surfactant protein D enhances clearance of influenza A virus from the lung in vivo. J. Immunol. 167, 5868-5873 (2001).
- Colamussi ML, White MR, Crouch E, Hartshorn KL. Influenza A virus accelerates neutrophil apoptosis and markedly potentiates apoptotic effects of bacteria. Blood 93(7), 2395-2403 (1999).
- Didierlaurent A, Goulding J, Patel S et al. Sustained desensitization to bacterial Toll-like receptor ligands after resolution of respiratory influenza infection. J. Exp. Med. 205(2), 323-329 (2008).
•• Provides important new insight into the pathogenesis of secondary bacterial infections after influenza virus infection. - McAuley JL, Hornung F, Boyd KL et al. Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe 2(4), 240-249 (2007).
• Provides fresh insight into bacterial superinfection. - Marsland BJ, Scanga CB, Kopf M, Le Gros G. Allergic airway inflammation is exacerbated during acute influenza infection and correlates with increased allergen presentation and recruitment of allergen-specific T-helper type 2 cells. Clin. Exp. Allergy 34(8), 1299-1306 (2004).
- Izurieta HS, Thompson WW, Kramarz P et al. Influenza and the rates of hospitalization for respiratory disease among infants and young children. N. Engl. J. Med. 342(4), 232-239 (2000).
- Dahl M, Dabbagh K, Liggitt D, Kim S, Lewis D. Viral-induced T helper 1 responses enhance allergic disease by effects on lung dendritic cells. Nat. Immunol. 5, 337-343 (2004).
- Tsitoura D, Kim S, Dabbagh K et al. Respiratory infection with influenza A virus interferes with the induction of tolerance to aeroallergens. J. Immunol. 165, 3484-3491 (2000).
- Strong P, Reid K, Clark H. Intranasal delivery of a truncated recombinant human SP-D is effective at down-regulating allergic hypersensitivity in mice sensitized to allergens of Aspergillus fumigatus. Clin. Exp. Immunol. 130, 19-24 (2002).
- Atochina EN, Beers MF, Tomer Y et al. Attenuated allergic airway hyperresponsiveness in C57BL/6 mice is associated with enhanced surfactant protein (SP)-D production following allergic sensitization. Respir. Res. 4(15), 1-12 (2003).
- Le Bon A, Durand V, Kamphuis E et al. Direct stimulation of T cells by type I IFN enhances the CD8+ T cell response during cross-priming. J. Immunol. 176(8), 4682-4689 (2006).
- Bracci L, Canini I, Venditti M et al. Type I IFN as a vaccine adjuvant for both systemic and mucosal vaccination against influenza virus. Vaccine 24(Suppl. 2), S2-56-57 (2006).
- Coro ES, Chang WL, Baumgarth N. Type I IFN receptor signals directly stimulate local B cells early following influenza virus infection. J. Immunol. 176(7), 4343-4351 (2006).
- Takeshita F, Tanaka T, Matsuda T et al. Toll-like receptor adaptor molecules enhance DNA-raised adaptive immune responses against influenza and tumors through activation of innate immunity. J. Virol. 80(13), 6218-6224 (2006).
- Fillatreau S, Manz RA. Tolls for B cells. Eur. J. Immunol. 36(4), 798-801 (2006).
- Tacchini-Cottier F, Zweifel C, Belkaid Y et al. An immunomodulatory function for neutrophils during the induction of a CD4+ Th2 response in BALB/c mice infected with Leishmania major. J. Immunol. 165(5), 2628-2636 (2000).
- Tezuka H, Abe Y, Iwata M et al. Regulation of IgA production by naturally occurring TNF/iNOS-producing dendritic cells. Nature 448(7156), 929-933 (2007).
- Zhou J, Matsuoka M, Cantor H, Homer R, Enelow RI. Cutting edge: engagement of NKG2A on CD8+ effector T cells limits immunopathology in influenza pneumonia. J. Immunol. 180(1), 25-29 (2008).
- Knudsen L, Ochs M, Mackay R et al. Truncated recombinant human SP-D attenuates emphysema and type II cell changes in SP-D deficient mice. Respir. Res. 8(1), 70 (2007).
- Palaniyar N, Clark H, Nadesalingam J et al. Innate immune collectin surfactant protein D enhances the clearance of DNA by macrophages and minimizes anti-DNA antibody generation. J. Immunol. 174(11), 7352-7358 (2005).
- Liu CF, Chen YL, Shieh CC et al. Therapeutic effect of surfactant protein D in allergic inflammation of mite-sensitized mice. Clin. Exp. Allergy 35(4), 515-521 (2005).
- Clark H, Palaniyar N, Strong P et al. Surfactant protein D reduces alveolar macrophage apoptosis in vivo. J. Immunol. 169, 2892-2899 (2002).
- Hickling T, Bright H, Wing K et al. A recombinant trimeric surfactant protein D carbohydrate recognition domain inhibits respiratory synctitial virus infection in vitro and in vivo. Eur. J. Immunol. 29, 3478-3484 (1999).
- Crouch E, Tu Y, Briner D et al. Ligand specificity of human surfactant protein D: expression of a mutant trimeric collectin that shows enhanced interactions with influenza A virus. J. Biol. Chem. 280(17), 17046-17056 (2005).
- Tecle T, White MR, Sorensen GL et al. Critical role for crosslinking of trimeric lectin domains of surfactant protein D in antiviral activity against influenza A virus. Biochem. J. (2008) (In Press).
- Tacken PJ, Hartshorn KL, White MR et al. Effective targeting of pathogens to neutrophils via chimeric surfactant protein D/anti-CD89 protein. J. Immunol. 172(8), 4934-4940 (2004).
- Zhang H, Porro G, Orzech N et al. Neutrophil defensins mediate acute inflammatory response and lung dysfunction in dose-related fashion. Am. J. Physiol. Lung Cell. Mol. Physiol. 280(5), L947-L954 (2001).
- Wu CC, Hayashi T, Takabayashi K et al. Immunotherapeutic activity of a conjugate of a Toll-like receptor 7 ligand. Proc. Natl Acad. Sci. USA 104(10), 3990-3995 (2007).
- Hammerbeck DM, Burleson GR, Schuller CJ et al. Administration of a dual Toll-like receptor 7 and Toll-like receptor 8 agonist protects against influenza in rats. Antiviral Res. 73(1), 1-11 (2007).
- Mazur I, Wurzer WJ, Ehrhardt C et al. Acetylsalicylic acid (ASA) blocks influenza virus propagation via its NF-κB-inhibiting activity. Cell. Microbiol. 9(7), 1683-1694 (2007).
- Welliver TP, Garofalo RP, Hosakote Y et al. Severe human lower respiratory tract illness caused by respiratory syncytial virus and influenza virus is characterized by the absence of pulmonary cytotoxic lymphocyte responses. J. Infect. Dis. 195(8), 1126-1136 (2007).
- Zheng B, Zhang Y, He H et al. Rectification of age-associated deficiency in cytotoxic T cell response to influenza A virus by immunization with immune complexes. J. Immunol. 179(9), 6153-6159 (2007).
- Deng Y, Jing Y, Campbell AE, Gravenstein S. Age-related impaired type 1 T cell responses to influenza: reduced activation ex vivo, decreased expansion in CTL culture in vitro, and blunted response to influenza vaccination in vivo in the elderly. J. Immunol. 172(6), 3437-3446 (2004).
- Boehmer ED, Goral J, Faunce DE, Kovacs EJ. Age-dependent decrease in Toll-like receptor 4-mediated proinflammatory cytokine production and mitogen-activated protein kinase expression. J. Leukoc. Biol. 75(2), 342-349 (2004).
- Seo S, Hoffman E, Webster R. Lethal H5N1 influenza viruses escape host anti-viral cytokine responses. Nat. Med. 8, 950-954 (2002).
- Salomon R, Hoffmann E, Webster RG. Inhibition of the cytokine response does not protect against lethal H5N1 influenza infection. Proc. Natl Acad. Sci. USA 104(30), 12479-12481 (2007).
•• Raises questions regarding the value of broad-based suppression of innate immune responses as a treatment strategy for pandemic influenza.
Authors and Disclosures
Mitchell R. White, Mona Doss, Patrick Boland, Tesfaldet Tecle, and Kevan L. Hartshorn, Boston University School of Medicine, Department of Medicine, EBRC 414, 650 Albany Street, Boston, MA, USA
E-Mail: Mitchell R. White - mitchw@bu.edu ; Kevan L. Hartshorn - khartsho@bu. edu
Disclosure: The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties. No writing assistance was utilized in the production of this manuscript.
Kevan L. Hartshorn, Boston University School of Medicine, EBRC 414, 650 Albany Street, Boston, MA 02118, USA. E-Mail: khartsho@bu. edu
Expert Rev Clin Immunol. 2008;4(4):497- 514. © 2008 Expert Reviews Ltd.
Ni Wikipedia ni los blogs que no contienen referencias pueden ser fundamento para tomar decisiones clínicas. Desgraciadamente el Dr. Gervas no especifica CON EVIDENCIAS y cientificamente si debe vacunar o no. Filosofa en base a principios y de alli llega a conclusiones. Asi llega a la conclusion que no hay que prevenir.
Saludos,
Jorge Chirinos
De: Carlos A. Morales P. <karlmoralesp@ yahoo.com>
Enviado: dom,20 junio, 2010 12:10
Asunto: [SALUD_LORETO] Quien es el Dr Juan Gérvas??????? ??
| Cuando los extranjeros hablan: Carlos Alberto Morales Paitán  Pediatra - Hospital del Niño - Lima, Perú - Mobile: 999-185-042 Acceso directo a mi Blog: www.karlmoralesp201 0.blogspot. com/ --- On Sun, 6/20/10, Carlos A. Morales P. <karlmoralesp@ yahoo.com> wrote:
|
No hay comentarios:
Publicar un comentario